Синдром ретта
Содержание:
Лечение патологии
К сожалению, на сегодняшний день технологии терапии генетических заболеваний находятся в стадии разработки. Поэтому для детей с болезнью Ретта применяется терапия сопутствующих проблем, снижение тяжелых симптомов и реабилитация. Комплекс мер позволяет обеспечить ребенку более качественную жизнь, не страдать от приступов эпилепсии и наладить работу внутренних органов.
Лекарственное лечение
В случае с данной патологией назначают следующие препараты:
- Ламотриджин или Карбамазепин, Финлепсин позволяют уменьшить частоту эпилептических припадков, снизят их силу.
- Ноотропные препараты простимулируют мозг. Например, Церебролизин.
- Во избежание периодов возбуждения нервной системы с выраженной капризностью, истериками назначают Глицин, Ноофен, Фенибут.
- Для установления нормального режима сна и бодрости применяют Мелатонин.
- Снятие спазмов, неконтролируемых движений и других паркинсонических проявлений обеспечат Перлодел, Бромокриптин.
- Также назначаются лекарства для лечения внутренних органов, системы ЖКТ, сердца, и прочее, что будет обнаружено при полном обследовании ребенка.
Реабилитация детей с синдромом Ретта
Обязательными мероприятиями считаются лечебная гимнастика и массаж, услуги мануального терапевта, но это далеко не все возможности изменить качество жизни больного к лучшему.
Физические упражнения
Развитие мозга ребенка во многом зависит от его двигательной активности. Поэтому упражнения помогают не только наладить движение, но и простимулировать ЦНС. Для каждого ребенка применяют индивидуальный комплекс упражнений, составленный специалистами по ЛФК. Некоторые могут выполняться с помощью специальных тренажеров, они позволяют научиться держать равновесие и ходить. Регулярное их выполнение существенно улучшат физическое состояние ребенка, его двигательные возможности, благотворно скажется на функциональности мозга.
Массаж
Действия мануального терапевта и просто массажиста направлены на снижение зажатости, гипертонуса мышц, формирование правильной осанки. Дети с болезнью Ретта нуждаются в периодических курсах мануального лечения и массажа.
Метод Томатиса
Прослушивание специальных музыкальных композиций по этой методике помогают нормализовать Психоэмоциональное состояние ребенка с синдромом, повышает его контактность.
Занятие с профильными специалистами
Психолог или психоаналитик поможет наладить контакт ребенка с родителем, подскажет, как правильно реагировать и отвечать на какие-то действия маленького пациента. Семья сталкивается с множеством психологических проблем с появлением малыша с тяжелой генетической патологией Ретта. Поэтому психотерапия проводится не только индивидуально с ребенком, но и со всеми членами этой семьи, чтобы помочь им создать благожелательную эмоциональную обстановку внутри своего дома.
Дефектологи, логопеды, и прочие специалисты наладят навыки общения такого пациента насколько это возможно.
Другие эффективные способы реабилитации
Существуют необычные методы реабилитации детей с генетическими патологиями, которые дают, в хорошем смысле, неожиданные результаты. К сожалению, они не входят в государственную программу для детей с синдромом, но могут применяться самостоятельно с согласования врача. Это:
- АВА-терапия. Специальные занятия вырабатывают навыки общения и поведения.
- Арт-терапия. Это не только рисование, но и другие творческие занятия, которые помогают эмоциональному развитию, обретению навыков движения ребенка.
- Иппотерапия – общение с лошадьми, а также верховая езда на специально подготовленных конях.
- Дельфинотерапия. Еще один метод реабилитации, основанный на общении с животными.
- Гидротерапия. Различные способы восстановления в воде. Ванны, души, гидромассаж, плавание, гимнастика и прочее очень эффективны при данной патологии.
- Канистерапия – общение и занятия с подготовленной собакой.
Продолжительность курса государственной реабилитации составляет месяц раз в полгода. Однако многое зависит от степени тяжести состояния пациента. Сроки может определить врач совместно со специалистом-реабилитологом.
Таким детям составляют специальную диету, которая предотвратит стремительную потерю веса при болезни Ретта и дефицит микроэлементов. Блюда выбирают высококалорийные, с достаточным содержанием витаминов и клетчатки. Интервалы между приемами пищи не должны превышать 3 часов. При плохом аппетите или отказе от еды, недостаток восполняют специальными смесями.
Что такое Синдром Ретта —
Прогрессирующее дегенеративное заболевание ЦНС предположительно генетического происхождения, встречается преимущественно у девочек, названо по имени австрийского ученого A. Rett, впервые описавшего его в 1966 г. Автор сообщил о 31 девочке с регрессией психического развития, аутистичным поведением, утратой целенаправленных движений и появлением особых стереотипных двигательных актов, «сжимания рук».
Распространенность
Частота его относительно высока — 1:10 000 девочек. В мире описано более 20 тыс. случаев заболевания; большинство из них спорадические, менее 1% — семейные. При изучении близнецов показана конкордантность по синдрому Ретта монозиготных и дискордантность дизиготных пар. Географическое распространение синдрома Ретта неравномерно. Отмечено скопление больных в определенных небольших сельских районах «Ретт-ареалы», что может быть связано с существующими популяционными изолятами. Такая концентрация заболевания наблюдается в Норвегии, Италии, Албании и Венгрии.
Истоки заболевания
В масштабном формате о расстройстве заговорили в 1983 году благодаря шведскому ученому Бенгту Хагбергу. В это время он со своей группой изучал 35 подобных между собой случаев в 3 разных странах: в Португалии, Франции и Швеции.
Однако Хагберт не является первооткрывателем синдрома. Впервые его обнаружил педиатр Андреас Ретт, имя которого носит заболевание. Он наблюдал за двумя девочками, имеющими одинаковые симптомы. Их он заметил в очереди на прием. Они сидели на коленях у матерей, а те держали их за руки. Девочки раскачивались как маятники, а затем внезапно обе начали совершать стереотипные движения руками. Дети застыли в одном положении, отстраненные от окружающего мира. Взгляд был направлен в одну точку. Поражала их синхронность в движениях и поведении.
В своих письменных архивах врач отыскал подобные истории болезни, а затем отправился в Европу, чтобы разыскать и там таких же пациентов. В 1966 он сделал первые публикации своих исследований, которые, однако, не вызвали особого интереса.
Зафиксированную им болезнь Ретт назвал синдромом атрофии мозга. Сначала ее считали проявлением аутизма или шизофрении, и только лишь в 1983 году вывели в отдельную нозологическую единицу.
В настоящее время синдром относят к категории довольно редких генетических заболеваний. Он встречается с частотой случаев 1 на 15000. Причиной его называют мутацию гена МЕСР2. Этот ген отвечает за синтез определенного белка, влияющего на развитие мозга. В норме этот белок, сп
устя некоторое время после рождения, должен подавляться другими генами, чтобы обеспечить нормальное развитие мозга.
Если же ген МЕСР2 мутирован, то белок инактивируется не полностью, что вызывает аномальное мозговое созревание, и провоцирует развитие синдрома Ретта.
Обычно мутирующий ген располагается в Х хромосоме, потому заболеванием страдают преимущественно девочки.
Прогноз
Чтобы найти адекватное лечение, медики и ученые по всему миру ведут интенсивные исследования детского заболевания, называемого «синдром Ретта». Данные и результаты научных центров, занимающихся этой проблемой, уже подтверждают версию о том, что процессы, которые запускаются патологией, обратимы.
Ведется активная разработка стратегии использования стволовых клеток, на которых будет основано лечение синдрома Ретта. Уже сегодня предварительные средства опробованы на лабораторных мышах. Опыты профессора Беличенко, которые проводились в Калифорнийском университете, дают положительный прогноз и надежду на скорое научное открытие эффективного лечения синдрома Ретта.
Специально предназначенных профилактических мер, предупреждающих развитие недуга, не существует, поскольку неизвестны причины синдрома Ретта. Для снижения вероятности появления на свет ребёнка с подобным диагнозом необходимо:
- вести здоровый образ жизни женщинам в период беременности;
- следить за адекватным протеканием гестации с регулярным посещением акушера-гинеколога;
- парам, решившим завести ребёнка, пройти консультацию у генетика.
Несмотря на то что подобное заболевание невозможно излечить, благодаря соблюдению рекомендаций лечащего врача можно достичь благоприятного прогноза – снижения интенсивности проявления симптомов, при этом продолжительность жизни человека может составлять 50 лет.
Тем не менее не следует забывать про высокую вероятность внезапной смерти больного. У детей летальный исход наиболее часто наступает из-за наличия сопутствующих патологий внутренних органов и полиорганной недостаточности. У взрослых смерть зачастую развивается на фоне инсульта.
Девочки способны прожить дольше в силу наличия дополнительной Х-хромосомы не подвергнутой мутационным изменениями. Без нормального гена, который способен обеспечить нормальные белки в дополнение к аномальным, что вызвано мутацией MECP2, XY-кариотип мужского пола не может замедлить развитие болезни. По этой причине, неспособность многих детей мужского пола с мутацией MECP2 выжить в перспективе проявляется наиболее ярко.
Взрослые женщины могут жить до 40 лет или более. Лабораторные исследования при синдроме Ретта у них могут показывать следующие аномалии:
- ЭЭГ нарушения от 2 летнего возраста, в процессе всей жизни;
- наличие атипичных гликолипидов в мозге;
- повышенные уровни бета-эндорфинов и глутамата;
- снижение субстанции Р;
- снижение уровня нервных факторов роста.
Доля смертности достаточно высока, но большинство летальных исходов не имеют определенных причин. В некоторых случаях смерть является результатом:
- спонтанной дисфункции ствола мозга;
- остановки сердца, вероятно, из-за длительного интервала QT-синдрома, желудочковой тахикардии или других типов аритмий;
- судорог;
- перфорации желудка.
На данный момент синдром Ретта не поддается лечению. Даже при наличии самой современной терапии ребенок не станет здоровым, не начнет реагировать на окружающие его предметы и не восстановит двигательную функцию. Но посредством современных методик можно снизить скорость деградации организма.
Причины возникновения синдрома Ретта

Этиология данной патологии довольно сложна не только для простого обывателя, но и для самих медиков. Основные причины синдрома Ретта можно озвучить следующими гипотезами:
- Мутация генов . Первоначально считалось, что именно кровосмешение (родственные связи) приводит к таким плачевным последствиям в виде прогрессирующего Х-сцепленного заболевания.
- Ломка Х-хромосомы (деформация проблемного участка в ее коротком плече) . Подобный патогенез способствует изменениям головного мозга, которые продолжаются до тех пор, пока ребенок не достигнет рубежа четырех лет.
- Дефицит нейротрофических факторов . Озвученное прерванное развитие вызывает деформацию нижних моторных нейронов, базальных ганглий, и в данный патологический процесс вовлекается спинной мозг.
То, что синдром Ретта имеет наследственный характер, — это факт. Однако среди специалистов до сих не существует единого мнения относительно природы образования озвученного Х-сцепленного заболевания.
Диагностические критерии синдрома Ретта
Несмотря на молекулярную генетику, синдром Ретта остается клиническим диагнозом. Диагностические критерии были обновлены в 2010 году2, Диагноз синдрома Ретта следует учитывать при постнатальном замедлении роста головы.
Типичный или классический синдром РеттаДля этого необходимо следующее:
- Период регрессии с последующим восстановлением или стабилизацией.
- Все основные критерии и все критерии исключения выполнены.
- Поддерживающие критерии не требуются, хотя они часто присутствуют при типичном синдроме Ретта.
Атипичный или вариантный синдром РеттаДля этого необходимо следующее:
- Период регрессии с последующим восстановлением или стабилизацией.
- Как минимум два из четырех основных критериев.
- Пять из одиннадцати поддерживающих критериев.
Основные критерииЭто:
- Частичная или полная потеря приобретенных целенаправленных ручных навыков.
- Частичная или полная потеря приобретенного разговорного языка.
- Нарушения походки: нарушение (диспраксия) или отсутствие способности.
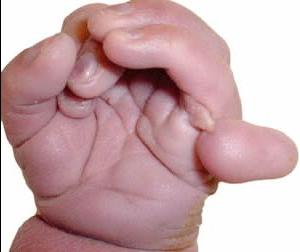
- Стереотипные движения рук, такие как автоматическое сжатие / сжатие, хлопки в ладоши, постукивание, полоскание рта и мытье / растирание.
Критерии исключения для типичного синдрома Ретта
- Повреждение головного мозга, вторичное по отношению к травме (перинатальной или постнатальной), нейрометаболическому заболеванию или тяжелой степени.
- Инфекция, которая вызывает неврологические проблемы.
- Грубое аномальное психомоторное развитие в первые шесть месяцев жизни.
Поддерживающие критерии для атипичного синдрома Ретта
- Нарушения дыхания при бодрствовании.
- Бруксизм, когда проснулся.
- Нарушение сна.
- Нарушение мышечного тонуса.
- Периферические вазомоторные нарушения.
- Сколиоз / кифоз.
- Ограничение роста.
- Маленькие холодные руки и ноги.
- Неуместные смеющиеся / кричащие заклинания.
- Уменьшается реакция на боль.
- Интенсивное глазное общение — «наведение глаз».
Поскольку мутации MECP2 в настоящее время выявляются в некоторых случаях до каких-либо явных признаков регрессии, диагноз «возможный» синдром Ретта следует ставить тем лицам в возрасте до 3 лет, которые не утратили никаких навыков, но в остальном имеют клинические признаки, свидетельствующие о синдроме Ретта. , Эти люди должны подвергаться повторной оценке каждые 6-12 месяцев для доказательства регрессии. Если регрессия проявляется, диагноз должен быть изменен на определенный синдром Ретта. Однако, если у ребенка нет признаков регрессии к 5 годам, диагноз синдрома Ретта следует поставить под сомнение.
Показано, что более ранний возраст диагноза приводит к тому, что семьи испытывают меньше стресса и эмоционального напряжения по сравнению с семьями с отсроченным диагнозом.4.
Особенности лечения
Поскольку заболевание спровоцировано нарушениями на генном и молекулярном уровнях, современная медицина не может полностью излечить страдающих синдромом Ретта. Однако существует комплекс мер и средств, направленный на хотя бы частичное улучшение состояния пациентов.
Основным (а на ранних стадиях – единственным) способом борьбы с недугом является медикаментозное лечение.
Чтобы блокировать приступы эпилепсии, таким детям прописывают «Карбамазелин» или любые другие сильнодействующие антиконвульсанты. Для установления стабильного режима сна используется препарат «Мелатонин».
Прописывают также препараты, улучшающие кровообращение и активизирующие мозговую деятельность – они не спасут от деградации полностью, но помогут замедлить ее и сделать не столь выраженной. Иногда доктора рекомендуют также «Ламотриджин» – новый препарат, ограничивающий проникновение в спинномозговую жидкость мононатриевой соли.
Ученые отметили, что у больных синдромом Ретта такое проникновение повышено, но они пока не могут точно сказать, является ли это причиной или следствием недуга (и влияет ли как-то на состояние пациента такое медикаментозное воздействие).
Однако не только лекарствами можно замедлить развитие синдрома Ретта. Врачи утверждают, что правильная диета также дает положительный результат; для его достижения в рационе следует увеличить содержание клетчатки, витаминов и продуктов с высоким содержанием калорий, поскольку все эти элементы жизненно необходимы для развития любого человеческого организма. Рекомендуется чаще кормить больного ребенка – возможно, хотя бы часть увеличенных порций пищи пойдет по назначению и поспособствует прогрессу малыша.
Для поддержания в исправном состоянии опорно-двигательного аппарата большое значение имеет специальный массаж, а также гимнастика. Если у здорового человека подобные упражнения вызывали бы явное развитие мышечной ткани, то у больного речь идет скорее о противостоянии деградации, но при такой проблеме даже такое достижение – уже неплохо.
Комплекс всех указанных мер не сделает ребенка здоровым, но позволит притормозить его деградацию, возможно – сохранить некоторые психоэмоциональные и двигательные способности, считающиеся нормальными для здорового человека. В крупных городах существуют центры для адаптации пациентов с тяжелыми нарушениями ментального характера, в которых занимаются и детьми с синдромом Ретта.
Остается только надеяться на то, что уже в ближайшее время ученые представят полноценное лекарство от этого недуга – или хотя бы еще на шаг приблизятся к облегчению состояния больных. Ведущие специалисты считают, что успешно противостоять развитию болезни можно будет с помощью вживления в организм искусственно выращенных стволовых клеток.
Сегодня ведется активная разработка такой технологии, поступают обрывочные сведения о том, что опытные образцы уже даже были испытаны на лабораторных мышах и дали положительный результат. Такие новости вселяют сдержанный оптимизм, но о точных сроках победы человечества над синдромом Ретта говорить пока не приходится.
В следующем видео вы можете посмотреть 1 выпуск цикла передач «Диагноз не меняет ребенка, он меняет вас», который ведет директор ассоциации синдрома Ретта.
Симптомы и признаки
Когда девочка с синдромом Ретта рождается, у нее нет никаких видимых нарушений. Базовые показатели — вес, общий внешний вид, окружность головы — в норме, а если и есть нарушения, то они не связаны с синдромом Ретта.
Первые три-четыре месяца ребенок развивается так же, как и его сверстники. На этом этапе отмечается только мышечная слабость, также наблюдаются следующие признаки:
- пониженная температура тела;
- выраженное потоотделение в зоне стоп и ладоней;
- бледность кожи.
Когда ребенок достигает полугода, у него отмечаются признаки отставания в процессах развития навыков движения (ползанье, переворачивание на спину). Он поздно начинает сидеть, стоять, не проявляет выраженного интереса к игрушкам, апатичен.
Признаки патологии на этапе младенчества могут отсутствовать и проявиться позднее.
Процесс прогрессирования синдрома Ретта делится на четыре стадии:
- I стадия. В самом начале жизни ребенок развивается обычными темпами, никаких серьезных нарушений в его развитии не отмечается. Возможно даже, что начальная стадия наступит спустя год-два после рождения: первая симптоматика синдрома Ретта наблюдается в промежутке между 6 месяцами и 2,5 годами жизни. Двигательное развитие ребенка замедляется, ему становится неинтересно играть, он безразлично относится к игрушкам и занятиям, которые ранее были интересны. Мышечный тонус падает, наблюдаются отклонения в физическом развитии: голова увеличивается медленнее, процесс удлинения трубчатых костей также замедляется, и ребенок выглядит ниже и слабее сверстников. Также могут возникнуть сердечно-сосудистые заболевания и сбои в функционировании органов желудочно-кишечного тракта.
- II стадия. Этот этап болезни наблюдается спустя 1-2 года после возникновения первых симптомов. Первыми признаками перехода во вторую стадию является возникновение нарушений сна (бессонница, частые пробуждения, некрепкий сон), ребенок выглядит тревожным. После этого дети стремительно, в течение нескольких недель, утрачивают почти все полученные ранее навыки, перестают ходить, разговаривать, не могут держать предметы в руках. На этом этапе возникают дыхательные нарушения: апноэ (прекращение дыхательной активности), длящееся на протяжении 1-2 минут, сменяется гипервентиляцией, когда ребенок глубоко и часто дышит. Эти отклонения в процессах дыхания наблюдаются, когда ребенок бодрствует, и исчезают в период сна.
Также наблюдаются и другие патологии: эпилептические приступы, нарушения координации, характерные для этого заболевания повторяющиеся движения, напоминающие процесс мытья рук.
III стадия. Процесс прогрессирования симптоматики замедляется. У детей отмечается тяжелая степень умственной отсталости, овладевание новыми навыками крайне затруднено, часто возникают эпилептические приступы, которые затруднительно контролировать. Также наблюдаются мышечные спазмы, непроизвольная двигательная активность (гиперкинезы). Но могут исчезнуть некоторые симптомы: улучшается сон, снижается уровень тревожности, ребенок выглядит более спокойным, взаимодействие с родителями и лечащими врачами облегчается. Этот период в среднем начинается в 2-4 года и завершается в 10-15 лет.
IV стадия. Количество эпиприступов сокращается, они могут исчезнуть полностью, но наблюдается стремительное прогрессирование двигательных отклонений. Большая часть детей на этом этапе практически полностью теряет способность двигаться, мышечная система атрофируется, возникают поражения сосудов ног, что увеличивает вероятность возникновения язв на фоне проблем с кровоснабжением. Сильно искривляется позвоночный столб из-за мышечной атрофии. Эта стадия длится всю оставшуюся жизнь.
Симптоматика, характерная для каждой из стадий, может меняться и тесно связана с индивидуальными особенностями ребенка и скоростью прогрессирования патологии.
Схожие с синдромом Аарскога расстройства
Симптомы следующих расстройств могут быть схожими с таковыми при синдроме Аарскога. Сравнения могут быть полезны для дифференциальной диагностики:
Синдром Нунана является относительно распространенным генетическим заболеванием, характеризующимся:
- низким ростом
- дисморфными чертами лица
- врожденным пороком сердца.
Расстройство характеризуется широким спектром симптомов и физических особенностей, которые сильно различаются по степени тяжести. У многих пораженных людей связанные нарушения включают:
- отличительный внешний вид лица;
- широкая или перепончатая шея;
- низкая задняя линия роста волос;
- типичная деформация груди
- низкий рост.
Характерные аномалии головы и лица (черепно-лицевой) области могут включать:
- широко расставленные глаза (глазной гипертелоризм);
- кожные складки, которые могут покрывать внутренние углы глаз (эпикантальные складки);
- опущение верхних век (птоз);
- маленькую челюсть (микрогнатия);
- подавленный носовой корень;
- короткий нос с широким основанием;
- низко посаженные назад повернутые уши.
Также обычно присутствуют отличительные пороки развития скелета, такие как аномалии грудины (грудины), искривление позвоночника (кифоз и/или сколиоз) и вальгусное колено. Многие дети с синдромом Нунана также имеют проблемы с сердцем, такие как обструкция правильного кровотока из нижней правой камеры сердца в легкие (стеноз легочных клапанов).
Дополнительные аномалии могут включать пороки развития определенных кровеносных и лимфатических сосудов, свертываемость крови и дефицит тромбоцитов, трудности в обучении или умеренную интеллектуальную неспособность, неспособность яичек опускаться в мошонку (крипторхизм) к первому году жизни у пораженных мальчиков и/или другие симптомы и признаки.
Синдром Нунана является генетически гетерогенным состоянием, которое может быть вызвано мутациями ряда генов PTPN11, KRAS, SOS1, RAF1, NRAS, RIT1 и SOS2.
Синдром Робинова — это редкое генетическое заболевание, наследуемое как доминантный и рецессивный признак и характеризующееся:
- невысоким ростом из-за задержек роста после рождения;
- отличительными аномалиями головы и лица;
- дополнительные пороки развития скелета;
- генитальные аномалии.
Черты лица у детей с синдромом Робинова напоминают черты лица восьминедельного плода; в медицинской литературе это состояние часто упоминается как «лицо плода».
Характерные черепно-лицевые особенности могут включать:
- аномально большую голову (макроцефалия) с выпуклым лбом;
- широко расставленные глаза;
- маленький вздернутый нос с ноздрями, расклешенными вперёд;
- затонувший (вдавленный) носовой мост.
Пороки развития скелета могут включать необычно короткие кости предплечья (брахимелия предплечья), аномально короткие пальцы рук и ног, постоянную фиксацию пятого пальца в согнутом положении (клинодактилия), необычно маленькие руки с широкими большими пальцами, порок развития ребра, сколиоз и/или недоразвитие одной стороны костей в средней (грудной) части позвоночного столба (гемивертебра).
Генитальные аномалии, связанные с синдромом Робинова, могут включать аномально маленький пенис (микропенис) и неспособность яичек опускаться в мошонку (крипторхизм) у пораженных мужчин и недоразвитие (гипоплазия) клитора и наружных удлиненных складок кожи с обеих сторон вагинального отверстия (половых губ) у пораженных женщин.
Диапазон и серьезность симптомов варьируются от случая к случаю.
Синдром Робинова является генетически гетерогенным состоянием, которое может быть вызвано мутациями в разных генах, таких как WNT5A, ROR2, DVL3 и DVL1.
Основные признаки
После 4–5 лет наблюдаются следующие проявления патологии:
- Постепенное замедление роста вплоть до полной остановки.
- Начавшаяся речевая активность затухает, способность общаться пропадает.
- Отставание в психическом развитии.
- Аномалия мышц ступней и ладоней.
- Возможно поредение волос.
- Уменьшение головы и мозга характерны для синдрома, это называется микроцефалией. При этом пропорции остального тела сохраняются.
- Не всегда ребенок может ходить, и даже если такая функция доступна, то только с помощью других людей.
- Тело скованно (гипертонус), расслабленность движения отсутствует при патологии Ретта. Либо, наоборот, мышцы вялые (гипотонус).
- Ребенок не может координироваться в пространстве.
- Непредсказуемые спазмы мышц – дистония. Они заставляют тело резко изменять свою позу.
- Приступы судорог. Эпилептические припадки.
- Патологии позвоночника в виде сколиоза и других нарушений.
- Болезни сердца, сбой ритма.
- Проблемы с органами ЖКТ.
- Лицо, мимика может искажаться от спазма мышц.
- Патологии дыхательной системы. Временная остановка, гипервентиляция легких.
- Бруксизм. Или по-другому ночной скрежет зубов.
- Глаза смотрят в одну точку или блуждают, взгляд расфокусированный.
Сочетание симптомов патологии Ретта различаются в зависимости от конкретного случая, зависят не только от степени поражения мозга, но и сколько предпринято мер по лечению и реабилитации.
Синдром Ретта у мальчиков
В основе синдрома лежит поражение гена MECP2, который расположен в Х-хромосоме. Именно благодаря этому гену в мозг поставляются белки и регулируется правильное развитие. И поэтому, когда повреждается данный ген, то появляется нарушение питания мозга, дефицит белка и поврежденные структуры мозга просто не могут его использовать.
У мальчиков всего одна Х-хромосома, поэтому они не могут компенсировать данное патологическое состояние, как девочки. Еще внутриутробно начинаются нарушения в развитии структур мозга, поэтому они умирают практически сразу же после рождения. Заболевание не успевает развиваться, и не проявляются симптомы, поэтому его тяжело диагностировать.
Если имеется также синдром Клайнфельтера, при котором у мальчика набор хромосом ХХУ. Именно благодаря второй Х-хромосоме новорожденному удается выжить. Вторым случаем, когда есть вероятность выживания, является не сильная выраженность мутации генов.
Диагностика заболевания
Диагноз ставит психиатр на основании опроса родителей и характерной клинической картины заболевания.
Международной ассоциацией по исследованию патологии были предложены диагностические критерии, разделённые на три группы: обязательные, второстепенные и исключающие.
К первой группе критериев относятся симптомы, которые могут свидетельствовать о классической форме синдрома. Обязательно наличие следующих признаков:
- беременность протекала нормально, в родах и послеродовом периоде осложнений не было;
- развитие до 5–18 месяцев было по возрасту;
- окружность головы при рождении соответствовала норме, замедление её роста началось в период от 5 месяцев до 4 лет;
- нарушение произвольных движений рук в период с полугода до двух с половиной лет;
- сильная задержка психомоторного развития, грубое нарушение речи;
- характерные стереотипии;
- расстройство координации движений, походки в возрасте 1–4 лет.
Дополнительные (второстепенные) признаки:
- нарушения дыхания;
- судорожные припадки;
- искривление позвоночника;
- задержка роста, в том числе гипотрофия ступней и кистей.
Исключающие критерии позволяют дифференцировать синдром Ретта с другими неврологическими патологиями. К ним относятся:
- внутриутробная задержка роста и другие аномалии развития;
- врождённая микроцефалия;
- перинатальное повреждение головного мозга;
- черепно-мозговые травмы;
- нейроинфекции.
Если есть хотя бы один такой признак, синдром Ретта исключается. Иногда набор симптомов не совсем соответствует классическому течению патологии. Тогда заболевание расценивают как неполную или атипичную форму.
Диагностические мероприятия начинаются с осмотра ребёнка психиатром и опроса родителей
Опросив родителей и осмотрев ребёнка, психиатр назначает дополнительные обследования:
- МРТ — позволяет выявить атрофию мозга лёгкой или средней степени, иногда атрофию мозжечка
- ЭЭГ (электроэнцефалограмма) регистрирует прогрессирующее замедление фоновой активности, изменения, характерные для эпилепсии.
Электроэнцефалограмма является дополнительным методом обследования ребёнка с синдромом Ретта
- Рентгенография стоп и кистей у большинства больных показывает укорочение локтевой, четвёртой плюсневой и пястной костей.
- УЗИ внутренних органов у четвёртой части больных показывает пороки развития селезёнки и печени.
- Методы современной генетики дают максимально полную диагностическую информацию, позволяя выявить мутацию гена, ответственного за развитие заболевания.
Дифференциальная диагностика
Дифференциальная диагностика главным образом проводится с детским нейрональным цероидным липофусцинозом (НЦЛ), детским церебральным параличом (ДЦП) и ранним аутизмом.
Для НЦЛ характерен стремительный регресс двигательных и коммуникативных навыков, этот диагноз исключается посредством проведения тканевого исследования электронным микроскопом.
Симптоматика синдрома Ретта очень сходна на раннем этапе с проявлениями аутизма:
- асоциальность ребёнка (нежелание контактировать с внешним миром и окружающими);
- нарушение речи;
- снижение способности к обучению; недержание кала и мочи;
- беспричинное беспокойство;
- повторяющиеся движения.
Таблица — дифференциальная диагностика с аутизмом
| Признаки | Синдром Ретта | Аутизм |
| Нормальное развитие до 6–18 месяцев | типично | не типично |
| Стереотипии | характерные движения руками | могут быть задействованы все части тела, движения разнообразные и сложные |
| Повторяющиеся манипуляции с различными предметами | не типично | повторяются постоянно |
| Моторика, координация движений, походка | прогрессирует апраксия и атаксия вплоть до полной неподвижности | нарушение общей и тонкой моторики, недостаточно координированная походка |
| Судорожный синдром | типичный признак | проявляется редко |
| Дыхательные нарушения | типичный симптом | не наблюдаются |
| Микроцефалия, гипотрофия стоп и кистей | всегда присутствуют | не типичны |
Диагностика синдрома Ретта
- Консультация невролога. Родители, которые стали замечать отклонения в развитии своего ребенка, должны обязательно посетить озвученного специалиста. Врач проанализирует жизненный анамнез малыша, оценит двигательные и речевые навыки юного пациента и измерит окружность его головы.
- КТ (компьютерная томография). После консультации у невролога может быть назначена данная процедура. С помощью этого метода диагностики синдрома Ретта производится анализ состояния головного мозга и фиксируется остановка его развития.
- ЭЭГ (электроэнцефалограмма). Если при расшифровке результата наблюдается медленный фоновый ритм, то это свидетельствует о наличии в организме мутации Х-хромосомы.
- Электрокардиография. У больных синдромом Ретта в несколько раз возрастает опасность возникновения инсульта. Следовательно, провести данную процедуру просто необходимо.
- УЗИ (ультразвуковое исследование). Подобный вид диагностики позволяет выявить неразвитость отдельных внутренних органов (печени, почек, селезенки и сердца) больного с синдромом Ретта. Именно по этой причине больные с таким диагнозом подвержены риску внезапной смерти.