Болезнь гоше
Содержание:
Патологическая анатомия
В разных органах обнаруживают мелкие или обширные очаги скопления клеток Гоше. Больше всего как при острой, так и при хронической форме болезни поражается селезенка. Она увеличена в размерах, нередко имеет бугристую поверхность. Ткань серо-красного, кирпичного или шоколадного цвета, на разрезе пестрая, с наличием ангиокавернозных очагов, инфарктов, рубцов. Микроскопически скопления клеток Гоше находят в красной пульпе, трабекулах, фолликулах; наблюдается резкое расширение синусов (но клеток Гоше здесь, как правило, нет). Клетки Гоше в селезенке содержат желто-коричневый пигмент. Печень также увеличена. Клеток Гоше здесь меньше, чем в селезенке; отдельные элементы представляют собой переходные формы от звездчатых эндотелиоцитов (купферовских клеток) к клеткам Гоше. Клетки Гоше располагаются диффузно в печеночных дольках, нарушая правильность их структуры, в стенках капилляров, вокруг синусов. В костном мозге очаговые скопления клеток Гоше сочетаются с рассасыванием костных балок, разрастанием соединительной ткани. В надпочечниках клетки Гоше находят преимущественно в ретикулярной зоне. В легких они инфильтрируют интерстициальную ткань альвеолярных перегородок, могут содержаться в просветах альвеол. При острой форме Г. б. эти клетки обнаруживают также в глиальных элементах головного мозга, тимусе, в клубочках почек. В головном мозге выявляют дистрофические изменения ганглиозных клеток, увеличение глиальных элементов в подкорковых узлах, явление нейронофагии (см.) и нарушение миелинизации. В белом веществе головного мозга иногда уменьшены отдельные фракции фосфолипидов, холестерина и цереброзидов, что может быть связано с демиелинизацией.
Рис. 1. Микропрепарат селезенки при болезни Гоше: видны округлые клетки Гоше (указаны стрелками).
Клетки Гоше (рис. 1) округлой формы, крупные (диам. 40—80 мкм) с небольшим ядром, часто располагающимся эксцентрично, и широкой зоной фибриллярной, исчерченной светло-серой цитоплазмы. Для клеток Гоше характерна интенсивно-голубая цитоплазма при окраске по методу Маллори (см. Маллори методы), положительная ШИК-реакция (см.), высокая активность кислой фосфатазы и неспецифической эстеразы. В цитоплазме нередко содержится гемосидерин. Встречаются многоядерные клетки. При электронной микроскопии в клетках Гоше и в клетках, еще не приобретших морфол. черт клеток Гоше, но уже накапливающих глюкоцереброзиды, находят отсутствующие в норме липидные цитосомы — образования в виде скопления трубочек, содержащих глюкоцереброзиды, диам. 25— 40 нм, отграниченные мембраной. При использовании метода негативного контрастирования установлено, что эти трубочки имеют спиралевидную структуру. Это отличает клетки Гоше от сходных с ними клеток, выявляемых при других заболеваниях, в частности при хроническом миелолейкозе (см. Лейкозы). При электронной микроскопии в клетках Гоше нередко обнаруживают и остатки эритроцитов.
Стандартные методы лечения
Основная цель терапии — улучшить качество жизни пациентов, позволяя им выполнять свою обычную повседневную деятельность, например, работать без ощущения чрезмерной усталости или нормально ходить, не испытывая боли в суставах. Другие цели включают предотвращение тяжелых осложнений, таких как снижение плотности кости до истончения, ослабление костей (остеопороз) и легких переломов или одышки из-за снижения функции легких. Нормализация роста ребенка для достижения им нормального роста также может быть целью терапии в течение нескольких лет лечения и достижения нормального начала полового созревания.
Лечение индивидуально для каждого пациента в зависимости от типа заболевания. Болезнь Гоше 1 типа считается излечимой и легкой, поскольку не включает неврологические симптомы, так как мозг не поражен. 2 тип не считается излечимым на данный момент из-за быстрого и необратимого повреждения головного мозга в младенческом возрасте. 3 тип все еще включает неврологическое повреждение, но эти симптомы прогрессируют медленнее, чем при 2 типе. Существуют современные FDA-одобренные варианты лекарственной терапии, которые включают ферментозаместительную терапию (ФЗТ) и субстрат-восстановительную терапию (СВТ).
Ферментозаместительная терапия (ФЗТ) оказалась эффективной для больных с 1 типом патологии. При исследованиях ФЗТ улучшалась анемия и низкий уровень тромбоцитов, значительно сокращалась увеличенная печень и селезенка, улучшались показатели скелета. Эти системные проявления также улучшаются у лиц с болезнью Гоше типов 2 и 3, которые проходят ФЗТ. Однако, ФЗТ не эффективен в терапии некоторых неврологических симптомов, связанных с болезнью Гоше типов 2 и 3.
ФЗТ предоставляется каждые 2 недели путем внутривенных инфузий либо в инфузионных центрах, в Национальном центре лечения болезни Гоше, либо на дому путем помощи члена семьи/друга или медсестры по уходу на дому. Три текущих одобренных FDA препарата ФЗТ включают имиглюцеразу (Церезим), альфа-велаглюцеразу (Вприв) и талиглюцеразу (Элисо).
Инъекция бесхозного лекарственного средства с алглюцеразой (Цередаза), являющаяся производным плаценты, была одобрена Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) в 1991 году для лечения болезни Гоше 1-го типа. Это была первая ФЗТ, эффективная для лечения заболевания 1 типа.
Синтетическая форма этого препарата, имиглюцераза (Церезим), была одобрена в 1994 году. Для получения Церезима используется технология рекомбинантной ДНК или генная инженерия. Это был важный шаг в преодолении ограничений доступности Цередазы, которая получалась из плаценты человека. Следовательно, Цередаза была изъята с рынка из-за производства аналогичных препаратов без проблем с биодоступностью из клеток, полученных от человека. Церезим, производимый Genzyme, заменяет человеческий лизосомальный фермент глюкоцереброзидазу, который отсутствует у людей с болезнью Гоше.
Другой одобренный FDA препарат глюкоцереброзидазы под названием альфа-велаглюцеразу (торговое название Вприв), полученный из непрерывной клеточной линии человека, производиться Shire.
Элисо (также известный как талиглюцераза альфа) от Pfizer Inc. по лицензии Protalix BioTherapeutics Inc., был одобрен FDA в 2012 году для лечения болезни Гоше 1 типа. Элисо представляет собой инъекционную длительную ферментозаместительную терапию, которая должна вводиться медицинским работником каждые две недели. Он использует генно-инженерные клетки моркови для замены глюкоцереброзидазы.
Субстрат-восстановительная терапия может также использоваться в определенных группах пациентов. Она работают иначе, чем ФЗТ, блокируя выработку глюкоцереброзида (жирного вещества) путем ингибирования фермента глюкозилцерамидсинтазы. Они входят в таблетки/капсулы и принимаются ежедневно. СВТ не должна использоваться у детей и подростков, беременных или кормящих женщин, пожилых пациентов и людей с тяжелыми заболеваниями почек или печени. Два текущих одобренных FDA лекарства включают Элиглустат и Миглустат.
В 2014 году Элиглустат, производимый Genzyme, был одобрен FDA для долгосрочного лечения взрослых пациентов с заболеванием 1-го типа.
В 2003 году Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США одобрило пероральную терапию Миглустатом для лечения взрослых пациентов с болезнью Гоше 1 легкой и средней степени тяжести, которые не могли воспользоваться ферментозаместительной терапией вследствие аллергии, гиперчувствительности и т.д…
Разновидности и причины заболевания
На сегодняшний день медики выделяют три основных типа заболевания, дифференцированные в зависимости от степени тяжести, возраста проявления симптомов и прогноза в лечении:
- самым распространенным типом с наилучшим прогнозом в лечении считается тип 1. Его симптомы редко проявляются в детском возрасте, зачастую это происходит на третьем десятке;
- второй тип считается самым редким. Проявления заболевания имеют место с самого рождения (зачастую все симптомы присутствуют по достижению полугодовалого возраста). Течение быстрое, а прогноз неутешительный – большая часть заболевших малышей не доживает и до двух лет;
- симптомы третьего типа болезни находят свое проявление в возрасте от 5 до 14 лет, прогноз также нельзя назвать благоприятным – практически все заболевшие погибают до совершеннолетия, и в редких случаях доживают до 30-ти.
Основная причина заболевания – это мутация глюкоцереброзида. Заболевание передается исключительно по наследству в том случае, если оба родителя являются носителями.
Болезнь Гоше может проявиться у детей, родители которых выглядят совершенно здоровыми
Считается, что каждый человек является носителем от 5 до 10 генов измененного типа. Некоторые изменения не оказывают на состояние организма особого влияния, а некоторые способны провоцировать развитие болезни. Риск появления болезни Гоше у ребенка одинаков как для девочек, так и для мальчиков. Аутосомно-рецессивный тип болезни свидетельствует о том, что заболеть ребенок может лишь при наследовании двух мутированных генов от обоих родителей.
Возможны и следующие варианты развития ситуации:
- когда один родитель не имеет подобной мутации, а второй является носителем болезни Гоше, болезнь развиваться не будет. Но при этом с вероятностью в 50% ребенок станет носителем заболевания;
- если оба родителя здоровы, то их дети получают не мутировавшие гены, а, следовательно, болезнь Гоше не может возникнуть вообще;
- когда один из родителей страдает от болезни Гоше в активной стадии, а второй не является ни заболевшим, ни носителем, все дети этой пары будут являться носителями, однако никто из них не заболеет;
- при условии носительства мутировавших генов обоими родителями, вероятность того, что ребенок будет носителем, составляет 50%, а заболевшим – 25%. Таким образом, у подобной пары вероятность рождения здорового ребенка все же есть;
- когда у одного родителя заболевание находится в активной фазе, а второй лишь носитель, оба варианта развития события (ребенок – заболевший и ребенок – носитель) имеют одинаковую вероятность в 50%.
Профилактика
В случае рождения в семье ребенка со злокачественной формой болезни при последующих беременностях матери больного показано исследование амниотической жидкости плода. При диагностике Гоше болезни у 11—17-недельного плода показано прерывание беременности. Специфической профилактики нет.
См. также Липоидозы, Сфинголипидозы.
Библиография: Берестов А. И. и Ковригин А. Е. К биохимической и цитохимической характеристике болезни Гоше у детей, Педиатрия, № 8, с. 33, 1972, библиогр.; Гусев Е.И. Клиническое и биохимическое изучение наследственных болезней обмена веществ с поражением нервной системы, Журн, невропат, и психиат., т. 71, № 10, с. 1475, 1971; Егоров П. И. и Мищенко А. С. Клиника, диагностика и лечение болезни Гоше, Педиатрия, № 8, с. 53, 1969, библиогр.; Жарко К. П., Митасова И. Н. и Ермакова Р.П. Болезнь Гоше у взрослых, Врач, дело, № 6, с. 143, 1969;
Покровский П. И. и Цепа Л. С. К вопросу о клинике, диагностике и терапии болезни Гоше, Пробл, гематол, и перелив, крови, т. 16, № И, с. 15, 1971, библиогр.; Рейнберг С. А. Рентгенодиагностика заболеваний костей и суставов, т. 1, с. 502, М., 1964; Bradу R. О., Jоhnsоn W. G. a. Uhlendorf B. W. Identification of heterosygous carriers of lipid storage disease, Amer. J. Med., v. 51, p. 423, 1971, bibliogr.; Brady R. O., KanferJ. N. a. Shapiro D. Metabolism of glucocerebrosides, Biochem. biophys. Res. Commun., v. 18, p. 221, 1965; Brooks S. E.H. a. Audretsch J. J. Ultrastructural diagnosis of Gaucher’s disease with negative-staining technique, Arch. Path., v. 95, p. 226, 1973; Cerebral sphingolipidoses, ed. by S. M. Aronson a. B. W. Volk, p. 73, N. Y. — L., 1962, bibliogr.; Danz M. u. Ka tenkamp D. Zur Gehirnbeteiligung beim Kongenitalen und friihinfantilen Morbus Gaucher, Zbl. allg. Path., Bd 115, S. 536, 1972; Gaucher P. E. De l’6pith61ioma primitif et isol6 de la rate, P., 1882; Goll E. У. u. Pexa H. Uber andauernde Ausschwemmung von Gaucher-Zellen ins Blut, Acta haemat. (Basel), Bd 31, S. 113,1964; Katz M. a. o. Maladie de Gaucher, J. Radiol. Electrol., t. 54, p. 61, 1973; The metabolic basis inherited disease, ed. by J. B. Stanbury a. o., p. 730, N. Y., 1972, bibliogr.; Miller J. D. Gaucher’s disease, Ann. intern. Med., v. 78, p. 883, 1973; Rosenfld-Striсhar d N. G. La maladie de Gaucher, 6tude d’une forme familiale, P., 1965, bibliogr.; Schneider E.L. a.o. Infantile (typ II) Gaucher’s disease, J. Pediat., v. 81, p. 1134, 1972, bibliogr.
Причины
До настоящего момента точная причина развития данной патологии до конца не выяснена, однако выделяются следующие предпосылки:
Это генетически обусловленная патология, которая наследуется по рецессивному принципу.
В результате мутации происходит нарушение строения участка гена, ответственного за ферментативную активность β-гликозидазы.
Данный фермент не может участвовать в реакции расщепления жиров.
В итоге жиры не перерабатываются в организме, а накапливаются в макрофагах клеточных структур различных органов и тканей.
Клетки видоизменяются, увеличиваются в размерах, перестают выполнять свои функции.
Это приводит к нарушению работы пораженных органов.
Риск заболеваемости возрастает в этнических группах населения, практикующих близкородственные браки.
Болезнь Гоше диагностика
Сегодня для диагностирования болезни Гоше используются разные методы.
Одним из самых точных считается анализ крови, при котором определяют наличие фермента и уровень глюкоцереброзидазы в лейкоцитых.
При анализе ДНК, втором методе, рассматривают на генетическом уровне мутации и недостающее количество основного фермента. Этот метод основан на новейших технологиях молекулярной биологии. Главное преимущество этой диагностики болезни Гоше состоит в том, что она позволяет проводить обследования на ранних сроках беременности. А это позволяет на 90%, в будущем, определить носителя заболевания и даже его степень тяжести.
При третьем способе диагностики исследуют костный мозг, что даёт возможность определить дисфункции его клеток. Отрицательная сторона такого обследования состоит в том, что оно позволяет диагностировать только больных, а выявить носителей заболевания оно не может. Поэтому на сегодня такой метод практически не применяется.
К сожалению, иногда симптомы болезни Гоше оказываются не всегда легкими. Поэтому именно правильная и своевременная диагностика может продлить больному жизнь на многие годы. При этом пациент будет находиться под наблюдением специалистов (педиатров, гематологов), не прибегая к использованию медикаментозного лечения и не опасаясь появления осложнений. И, напротив, если больной, который своевременно не прошёл квалифицированную диагностику и не получил медицинскую помощь, может на протяжении всей жизни иметь симптомы болезни, страдать от них и не догадываться, что у него болезнь Гоше, соответственно, и не получить правильного лечения.
Описание
Болезнь Гоше — это редкое наследственное лизосомное заболевание накопления, развивающееся в результате недостаточности бета-гликозидазы. Недостаточность данного ферментативного комплекса приводит к накоплению глюкоцереброзида в макрофагах и отложению его в таких органах, как селезёнка, головной и костный мозг, лёгкие, печень.
Заболевание открыто в 1882 французским врачом-дерматовенерологом Филиппом Гоше.
Классификация болезни Гоше
Международная классификация заболеваний (МКБ-10) относит болезнь Гоше к классу E75.2 и в группу заболеваний, связанных с нарушением обмена сфинголипидов.
Виды болезни Гоше:
- 1 тип, или нейронопатическая форма болезни Гоше — встречается с частотой примерно 1:20000-1:55000 населения в зависимости от этнической группы. Например, среди ашкенази частота встречаемости составляет 1:25000 человек. Хоть заболевание и отмечается практически у всех этнических групп, однако чаще наблюдается у еврейского населения. I тип заболевания проявляется уже у детей первых нескольких лет жизни и сопровождается увеличением печени, селезёнки (с возможными подкапсульными гематомами и разрывами). Появляются нарушения в работе опорно-двигательной системы с преобладанием поражения костных структур. При болезни Гоше развивается анемия, тромбоцитопения. В результате тромбоцитопении появляются частые гематомы, а в результате анемии развивается хроническая усталость. При лёгком течении заболевания клиническая симптоматика может не проявляться. Поражение костной системы у взрослых и детей при I и III типе является одним из главных факторов инвалидизации.
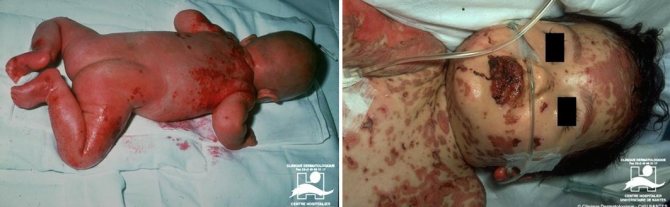
- 2 тип, или инфантильно-нейронопатическая форма болезни Гоше — чаще встречается у нееврейского этнического населения. Частота встречаемости составляет 1:90000 новорожденных. II тип развивается в первые дни после рождения и может проявляться вплоть до 18-летнего возраста — чаще в возрасте 3,5 месяцев. К 6 месяцам у детей появляются признаки умственной неполноценности, судорожного синдрома, апноэ. Помимо увеличенной печени и селезёнки (как и при первом типе) наблюдается поражение головного мозга, ригидность верхних и нижних конечностей, нарушенная моторика глаз. Прогноз при II типе болезни Гоше неблагоприятный, дети умирают примерно в возрасте 1,5 лет в связи с рецидивирующими пневмониями в результате прогрессирующей бульбарной дисфункции.
- 3 тип, или ювенильная нейронопатическая форма болезни Гоше — клиническая симптоматика начинает появляться у детей в возрасте 2-3 лет. Начинается всё с увеличения селезёнки, при этом увеличение печени менее выражено. После появления спленомегалии заболевание не проявляет себя около 5-6 лет, а затем начинают появляться неврологические симптомы. Прогноз при III типе болезни Гоше умеренно благоприятный в плане выживаемости (пациенты доживают до достаточно взрослого возраста), однако неблагоприятный в случае с инвалидизацией (в связи с поражением костных структур 3 и 1 типы заболевания приводят к ранней инвалидизации человека).
Осложнениями при болезни Гоше являются непосредственно клинические симптомы, которые возникают в том или ином возрасте. То есть осложнением течения заболевания можно считать возникновение неврологических синдромов, деформации, гематомы и другие.
Образ жизни при болезни Гоше существенно изменяется у пациентов, которых доживают до сознательного возраста. Это связано не только с поражением опорно-двигательного аппарата, но и с задержкой умственного и психического развития.
Какая вероятность передачи болезни Гоше ребенку от родителей?
Болезнь Гоше может передаться ребенку с вероятностью 25% от мужчины и женщины, которые являются носителями болезни Гоше. При этом у них нет самого заболевания, но каждый имеет один дефектный ген GBA. При рождении у них ребенка имеется следующая вероятность рождения детей:
- Имеется 1 шанс из 4 (т.е. вероятность 25%) того, что ребенок унаследует 2 нормальных гена GBA и у него не будет заболевания (он не будет ни больным с болезнью Гоше, ни ее носителем)
- Имеются 2 шанса из 4 (т.е. вероятность 50%), того, что ребенок унаследует 1 дефектный ген GBA и будет носителем болезни Гоше (но у него будут отсутствовать нарушения, вызванные заболеванием)
- Имеется 1 шанс из 4 (т.е. вероятность 25%) того, что ребенок унаследует 2 дефектных гена GBA и у него будет болезнь Гоше
- Если только один из родителей является носителем болезни Гоше, то у ребенка НЕ будет заболевания, но ребенок может быть носителем болезни Гоше.
- Если оба родителя не являются носителями дефектного гена, их дети не будут иметь болезнь Гоше и не будут ее носителями.
Схема наследования болезни Гоше.
Оба родителя являются носителями болезни Гоше
Справочная литература
- Genetic Alliance, The New York-Mid-Atlantic Consortium for Genetic and Newborn Screening Services. Understanding Genetics: A New York, Mid-Atlantic Guide for Patients and Health Professionals. Washington (DC): Genetic Alliance; 2009 Jul 8.
- Grabowski GA, Petsko GA, Kolodny EH. Chapter 146: Gaucher disease. Valle D, Beaudet AL, Vogelstein B, et al, eds. The Online Metabolic and Molecular Bases of Inherited Disease. New York, NY: McGraw Hill; 2013. http://ommbid.mhmedical.com/content.aspx?bookid=474§ionid=45374148. Accessed July 6, 2015.
История открытия болезни
В 1882 году, будучи еще студентом, Филипп Гоше заметил, что у его пациентки в возрасте 32 лет неестественно крупная селезенка. В итоге больная скончалась от сепсиса. Был проведен тщательный осмотр всех органов женщины, и студент заметил, что объем селезенки и печени в разы превышает норму. Он подумал, что это одна из разновидностей рака селезенки. Но симптомы описал очень подробно, именно его исследование помогло ученым 20 века выявить истинную причину болезни. Поэтому заболевание и назвали именем Гоше.
В 20 веке ученые рассмотрели в клетках пораженной селезенки глюкоцереброзид – тот самый продукт накопления, жир. И только в 1965 г. стало понятно, что скапливается жир из-за неработающего глюкоцереброзидаза – расщепляющего фермента. Он препятствует нормальному функционированию большинства внутренних органов.
Современной науке известно множество форм мутаций, которые отрицательно сказываются на образовании в организме фермента. Именно это является причиной слишком разнообразных и появлению все новых симптомов болезни Гоше.
Разновидности болезни Гоше
Фото больных представлено в статье. В современной медицине наблюдается три основных вида этого заболевания. Но, к сожалению, в полной мере причины болезни Гоше не изучены.
- Первый тип — является наиболее распространенным среди остальных и встречается примерно у 50 человек из 70 000. У некоторых пациентов он может протекать спокойно, не вызывая яркой симптоматики, у других же — возможно возникновение очень серьезных нарушений, которые нередко становятся угрожающими их жизни. В данном случае начинается процесс поражения мозга и нервной системы.
- При втором типе наследования болезнь Гоше имеет симптомы выраженной нейронопатии. Встречается крайне редко, примерно в одном случае на 100 000 человек. Симптоматика этой разновидности болезни Гоше наблюдается уже на первом году жизни. При этом у ребенка развиваются серьезные неврологические нарушения. По статистике, такие дети не доживают до трех лет.
- Третий тип характеризуется развитием хронической формы нейронопатии, встречается так же редко, как и болезнь 2-го типа. В данном случае наблюдается ярко выраженная неврологическая симптоматика, однако болезнь протекает более спокойно. Проявляются симптомы в раннем детстве, но, тем не менее, человек может дожить до зрелости.
К каким докторам следует обращаться если у Вас Болезнь Гоше (глюкоцереброзидный липидоз, глюкоцереброзидоз):
Генетик
Педиатр
Вас что-то беспокоит? Вы хотите узнать более детальную информацию о Болезни Гоше (глюкоцереброзидного липидоза, глюкоцереброзидоза), ее причинах, симптомах, методах лечения и профилактики, ходе течения болезни и соблюдении диеты после нее? Или же Вам необходим осмотр? Вы можете записаться на прием к доктору – клиника Eurolab всегда к Вашим услугам! Лучшие врачи осмотрят Вас, изучат внешние признаки и помогут определить болезнь по симптомам, проконсультируют Вас и окажут необходимую помощь и поставят диагноз. Вы также можете вызвать врача на дом. Клиника Eurolab открыта для Вас круглосуточно.
Как обратиться в клинику:
Телефон нашей клиники в Киеве: (+38 044) 206-20-00 (многоканальный). Секретарь клиники подберет Вам удобный день и час визита к врачу. Наши координаты и схема проезда указаны здесь. Посмотрите детальнее о всех услугах клиники на ее персональной странице.
Лечение болезни Гоше
Если у пациента нормальное общее состояние, селезенка увеличена незначительно, почти отсутствует анемия, то лечение болезни Гоше не проводят.
В случае резкого увеличения селезёнки, наличии изменений в костном мозге, сильной кровоточивости единственным результативным методом лечения болезни Гоше является удаление селезёнки.
Удаление селезенки может быть также заменено на заместительную ферментотерапию, которая заключается во введении пациенту один раз в 14 дней недостающего или отсутствующего фермента. Такой метод лечения применяется только для пациентов с тяжелыми симптомами болезни Гоше.
Для проведения такой терапии применяется имиглюцераза – фермент, который был получен в результате применения генно-инженерных технологий. Целью лечения болезни Гоше имиглюцеразой является предотвращение необратимых изменений в костно-суставной системе и остальных органах, уменьшение размеров селезенки и печени, ослабление цитопении. Препарат вводится пациентам внутривенно. В некоторых случаях лечение может назначаться пожизненно.
Контроль за результативностью проведенной заместительной ферментотерапии заключается в отслеживании показателей общего анализа крови (проводится раз в квартал), биохимического анализа крови (раз в полгода), определении размеров печени и селезенки, оценке состояния костей и суставов.
Также в лечении применяется гормональная терапия, рентгентерапия селезёнки с введением препаратов, стимулирующих костномозговое кроветворение.
Если выраженное селезеночное увеличение отсутствует, нет явных признаков остеопении, применяют лечение гепактопротекторами, желчегонными препаратами, препаратами кальция, витамином D.
Тяжелые случаи требуют проведения терапии сдерживания, включающей низкие дозы химиопрепаратов.
Если развиваются необратимые повреждения костей и суставов, то больным показано ортопедическое лечение.
Болезнь Гоше является довольно редким генетическим заболеванием, развивающимся по причине недостаточности в организме такого фермента, как глюкоцереброзидаза, что ведет к накоплению в различных органах и тканях глюкоцереброзида.
Проявляется болезнь увеличением селезенки и печени, анемией, тромбоцитопенией, слабостью костей и выраженными костными заболеваниями. Болезнь имеет неоднозначные внешние проявления, отличающиеся у разных пациентов, что затрудняет постановку диагноза.
Для диагностики данного заболевания применяют молекулярный анализ для определения уровня глюкоцереброзидазы, исследование костного мозга и другие методы, в том числе рентгенографию, МРТ, денситометрию.
Основным методом лечения болезни Гоше сложных форм является заместительная ферментная терапия, от своевременности назначения которой зависит прогноз для жизни пациента.