Синдром арнольда-киари
Содержание:
Аномалия Кимерли (Киммерли, Kimmerle)
Задние отделы мозга питаются, главным образом, из системы позвоночных артерий, проходящих в полость черепа прямо через шейные позвонки. Позвоночные артерии (правая и левая) огибают первый шейный позвонок, затем проходят в полость черепа, где и включаются в общую систему мозгового кровообращения.
При аномалии Кимерли над дугой первого шейного позвонка имеются аномальные дополнительные костные дужки-полукольца, которые могут сдавливать позвоночные артерии. Нарушение притока крови к мозгу может выявлено при ультразвуковом исследовании сосудов.
Сочленение черепа с первым шейным позвонком (рентген) в норме и при аномалии Кимерли (Киммерли). Первый шейный позвонок огибает позвоночная артерия, на рисунках она изображена красным цветом. При аномалии Кимерли над артерией видна аномальная дужка-полукольцо – виновница ущемления артерии. Эту же дужку видно и на рентгеновском снимке.
Аномальные дужки-полукольца хорошо видны на обычных рентгеновских снимках, однако врачи рентгенологи не всегда распознают наличие упомянутых дужек, поэтому врачи нашей клиники читают снимки самостоятельно.
Ущемление артерий с нарушением мозгового кровообращения можно увидеть с помощью УЗИ сосудов головного мозга, при этом информативны пробы с различными поворотами и наклонами головы. При аномалии Кимерли при УЗИ можно увидеть, как в одном или нескольких положениях головы кровоток по пострадавшей артерии резко обедняется.
Обычно артерии сдавливаются между аномальной дужкой и чрезмерно напряженными (спазмированными) мышцами шеи. Сдавление позвоночных артерий приводит к дефициту кровоснабжения головного мозга, отсюда и неврологические симптомы. Восстановления нормального тонуса и подвижности мышц шеи обычно приводит к восстановлению нормального самочувствия при аномалии Кимерли. .
При аномалии Кимерли мы, как правило, наблюдаем два-три или более из перечисленных симптомов:
- Головокружение и/или шаткость (может усиливаться при повороте головы);
- Шум (звон, гул, свист, шипение т.п.) в одном или обоих ушах (может усиливаться при повороте головы);
- Потемнение в глазах, “мушки” или “звездочки” перед глазами при напряжении или некомфортном положении мышц шеи, повороте головы;
- Утрата сознания (полная или частичная) глазами при напряжении или некомфортном положении мышц шеи, повороте головы;
- Внезапная слабость мышц и падение при сохраненном сознании.
В более тяжелых случаях возможно следующее:
- Головная боль;
- Нистагм (непроизвольное подергивание глазных яблок);
- Тремор рук, ног, расстройств координации движений;
- Снижение чувствительности части лица, части туловища, одной или нескольких конечностей;
- Слабость мускулатуры части лица, части туловища, одной или нескольких конечностей.
В некоторых случаях возможно развитие состояний, угрожающих инсультом головного мозга.
Симптоматика
Аномалия Киари I типа — самая распространенная форма синдрома, клинические признаки которой условно объединены в пять синдромов:
- Гипертензионный синдром проявляется цефалгией, подъемом артериального давления в утренние часы, напряженностью и гипертонусом шейных мышц, дискомфортом и болезненными ощущениями в шейном отделе позвоночника, диспепсическими явлениями, общей астенизацией организма У новорожденных детей возникает общее беспокойство, рвота фонтаном, тремор подбородка и конечностей, нарушается сон. Ребенок постоянно плачет, отказывается от груди.
- При наличии мозжечковых нарушений у больных изменяется произношение, речь становится скандированной, возникает вертикальный нистагм. Они жалуются на частые головокружения, рассогласованность движений, шаткость походки, дрожание рук, нарушение равновесия, дезориентацию в пространстве. Больные с большим трудом выполняют простые целенаправленные действия, в движениях отсутствует четкость и скоординированность.
- Поражение черепно-мозговых нервов проявляется признаками корешкового синдрома. У пациентов ограничивается подвижность языка и мягкого неба, что приводит к нарушению речи и проглатывания пищи. Их голос изменяется в сторону гнусавости и осиплости, речь становится неясной, дыхание затрудненным. Нарушение ночного дыхания отмечаются у большинства больных. У них возникает гипопноэ, центральное или обструктивное апное, при прогрессировании которого развивается острая дыхательная недостаточность. Лица с синдромом плохо слышат и видят, у них двоится в глазах и шумит в ушах. Со стороны органов зрения пациенты отмечают наличие светобоязни и боль при движении глазными яблоками. Офтальмологи часто обнаруживают анизокорию, спазм аккомодации или скотомы. Одним из основных симптомов синдрома является гипестезия – снижение чувствительности кожи лица и конечностей. Подобные патологическое изменения связаны с приглушенным реагированием рецепторов кожи на внешние раздражители: тепло или холод, уколы, удары. В тяжелых случаях нервное окончания вообще перестают воспринимать различные экзогенные воздействия.
- Сирингомиелический синдром — сложный симптомокомплекс, проявляющийся парестезией или онемением конечностей; изменением тонуса мышц и их гипотрофией, приводящей к миастеническим расстройствам; поражением периферических нервов, проявляющимся болью в конечностях; дисфункцией органов таза в виде затрудненной дефекации или самопроизвольного мочеиспускания; возможны артропатии — поражения суставов.
- У больных с пирамидальной недостаточностью снижается сила в нижних конечностях и способность к тонким движениям, ограничивается объем движений, повышается мышечный тонус — так называемая спастичность, например, спастическая походка. Повышение сухожильных рефлексов сочетается с одновременным снижением кожных рефлексов — брюшных. Возможно появление патологических рефлексов. У пациентов страдает мелкая моторика рук.
Любое неосторожное движение усиливает симптомы патологии, делает их более выраженными и яркими. Изменение положения головы — частая причина потери сознания
Этиология
Причины синдрома в настоящее время точно не определены. Существует несколько теорий происхождения патологии, но до конца ни одна из них не имеет официального подтверждения. Мнения ученых-неврологов всего мира до сих пор расходятся.
Большинство медиков признают синдром врожденным недугом, сформированным в процессе эмбриогенеза под воздействием негативных факторов внешней среды, оказывающих свое пагубное действие на женский организм при беременности. К ним относятся: самостоятельные использование лекарств, алкоголизм, табакокурение, вирусные инфекции, ионизирующее излучение.

Врожденные причины:
- Дистопия мозжечковых миндалин за пределы задней черепной ямки, имеющей относительно малые размеры;
- Выталкивание растущих структур мозга через затылочное отверстие;
- Неправильное формирование в процессе эмбриогенеза и атипичное развитие в постнатальном периоде костной ткани, приводящее к деформации черепной коробки.
Некоторые ученые определенную роль в развитии синдрома отводят генетическому фактору. В настоящее время можно точно утверждать, что болезнь не связана с хромосомными аномалиями.
Другие ученые придерживаются иной точки зрения относительно происхождения синдрома. Они считают его приобретенным и объясняют свое мнение появлением симптомов патологии у взрослых лиц. Приобретенный синдром возникает под действием экзогенных факторов. У больного новорожденного ребенка череп может иметь нормальное строение без костных аномалий и гипоплазии.
Приобретенные причины:
- Родовой травматизм с поражением черепа и мозга,
- Воздействие цереброспинальной жидкости на стенки спинного мозга,
- Любые ЧМТ,
- Бурный рост мозга в условиях медленно растущих костей черепа.
Симптомы патологии долгое время отсутствуют у больных, а затем внезапно появляются под воздействием провоцирующих факторов: вирусов, травм головы, стрессов.
Опущение основных мозговых структур до шейных позвонков блокирует процесс перетекания ликвора из подпаутинного пространства в спинномозговой канал. Это приводит к дисциркуляторным изменениям. Ликвор, продолжая синтезироваться и никуда не оттекая, скапливается в головном мозге.
Ученый Киари в 1891 году выделил четыре типа аномалии:
I – выход структурных элементов головного мозга за пределы задней черепной ямки, обусловленный недоразвитием костной ткани этой области. Этот тип клинически проявляется у лиц зрелого возраста.
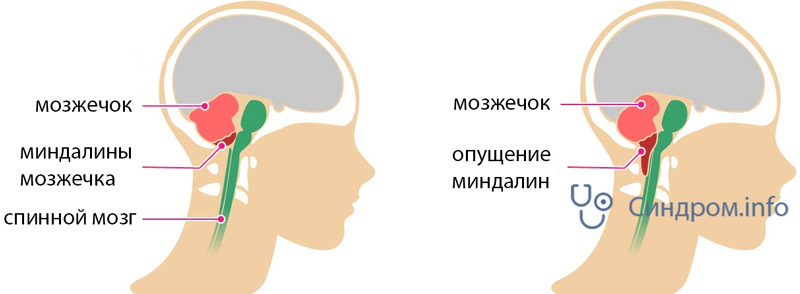
выход структур мозжечка за пределы ЗЧЯ при аномалия 1 типа
II — нарушения эмбриогенеза, приводящие к расположению структур мозжечка и продолговатого мозга ниже большого затылочного отверстия.
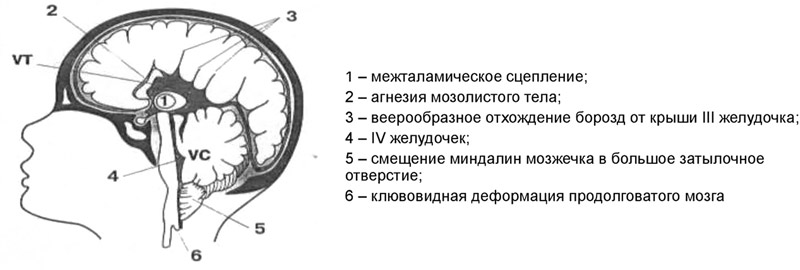
синдром Арнольда-Киари II типа
III — эктопия мозговых структур в каудальном направлении с образованием энцефаломенингоцеле.
аномалия III типа
IV — недоразвитый мозжечок не смещается и не выходит за пределы черепа. Поскольку отсутствует грыжевое выпячивание мозга, этот тип синдрома отсутствует в современной классификации.
Существуют два новых типа синдрома. Тип — мозжечок располагается достаточно низко, но при этом находится в черепной коробке. Тип 1.5 – промежуточная форма, сочетающая признаки I и II типов.
Выделяют три степени тяжести патологии:
- Первая — относительно легкая форма патологии без аномалий мозговых структур и характерных клинических проявлений.
- Вторая — наличие пороков развития ЦНС с врожденным недоразвитием головного мозга и подкорки.
- Третья — аномалии строения головного мозга со смещением мягких тканей в отношении твердых структур, образованием ликворных кист и сглаженностью извилин.
Что такое синдром Арнольда-Киари?
В задней черепной ямке в норме располагаются такие важнейшие структуры, как часть полушарий, продолговатый мозг, миндалины мозжечка. В этой области присутствует переход головного мозга в спинной. Структуры спинного мозга переходят в область шейного позвоночного канала через затылочное отверстие.
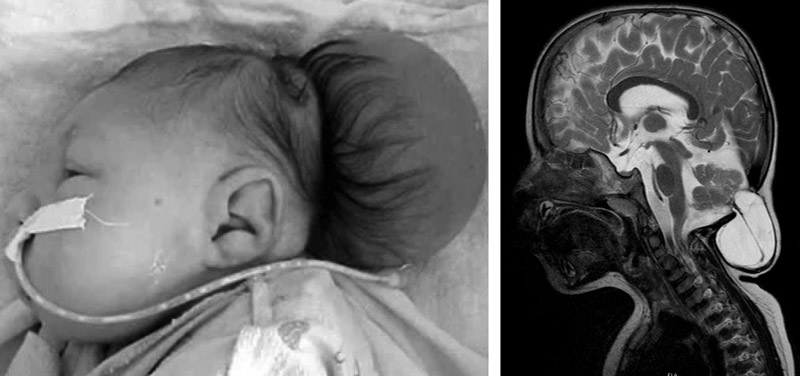
Мальформация Киари характеризуется появлением нарушений развития костей черепа. Затылочная часть увеличена у ребенка в этом случае редко. Это патологическое состояние возникает на фоне внутриутробной гидроцефалии.
Нарушения в строении черепной коробки приводят к тому, что структуры мозга смещаются вниз. Формируется своеобразная грыжа. Может произойти компрессионное повреждение структур мозга, что влечет за собой тяжелые нарушения работы основных центров, регулирующих работу всех систем организма. Может произойти сдавливание спинного мозга. Смещение структур головного мозга может быть выявлено при проведении миелоэнцефалографии.
Типы и степени аномалии Арнольда-Киари
В зависимости от степени выраженности изменений со стороны костного основания черепа и структур костного мозга выделяют 4 типа течения патологии.
Наиболее часто встречающейся и имеющей относительно благоприятный прогноз является мальформация Киари Первого типа. Это форма течения патологии отличается смещением структур миндального тела и мозжечка вниз.
Мальформация Арнольда-Киари Второго типа начинает проявляться в раннем детском возрасте. В этом случае присутствует смещение большего количества мозговых структур и наличие дополнительных пороков развития.
При синдроме Арнольда-Киари Третьего типа наблюдается выпячивание не только моста, продолговатого мозга, миндалевидного тела, но и мягкой мозговой оболочки. Патология приводит к тяжелым неврологическим нарушениям.
При синдроме Арнольда-Киари Четвертого типа наблюдается недоразвитие мозжечка, передавливание спинного мозга и критическое смещение структур головного. Такие новорожденные дети являются нежизнеспособными.
Выделяются 3 степени тяжести синдрома Арнольда-Киари. Относительно легким вариантом патологии, при котором не наблюдается развития тяжелых пороков головного мозга и выраженных симптоматических проявлений, считается синдром Арнольда-Киари 1 стадии.
Более опасными считаются 2 и 3 степени синдрома. Они сочетаются с другими врожденными нарушениями формирования тканей нервной системы. Это ухудшает прогноз. Даже при направленном лечении нередко у детей наблюдаются тяжелые неврологические нарушения, которые почти не поддаются коррекции.
Причины и механизм развития порока задней черепной ямки
Точные причины развития этой аномалии еще не установлены. Считается, что проблема кроется в генетических сбоях при развитии плода, ведущих к неправильному формированию структур головного мозга и костей черепа.
Многие специалисты считают, что проблема может быть обусловлена наличием врожденной гидроцефалии.
Накапливающаяся жидкость в период внутриутробного развития увеличивает давление внутри мозга, приводя к выдавливанию его структур в затылочное отверстие.
К факторам, повышающим риск развития патологии, относятся:
- черепно-мозговые травмы во время родов;
- бесконтрольный прием препаратов;
- пристрастие к алкогольным напиткам и табакокурению;
- вирусные инфекции.
Особенности болезни
Несмотря на то, что характерные признаки заболевания могут появиться уже во взрослом возрасте, этот дефект является врожденной патологией. В большинстве случаев она развивается в результате нарушения формирования костей черепа. Из-за этого у пациента может быть выявлен аномально маленький размер черепной ямки. Повышает риск появления подобной проблемы расширенная форма затылочного отверстия черепа.
Терапия патологии
Лечение заболевания, которое протекает бессимптомно, не проводится. Если у пациентов в области затылка или шеи наблюдается болезненности и при этом отсутствует другая симптоматика, то проводится консервативная терапия. Пациентам рекомендован прием миорелактирующих, анальгезирующих и противовоспалительных лекарств.
При наличии неврологических нарушений в виде расстройств чувствительности, парезов, нарушения тонуса мышц, а также болезненности, которую невозможно купировать препаратами, назначается хирургическое вмешательство.

Хирургическое вмешательство, при которых задние половины двух первых позвонков шеи и мозжечок поддается резекции, позволяет ликвидировать сдавливание ствола. В ходе операции подшивается заплата из искусственного материала в твердую мозговую оболочку, что нормализует циркуляцию цереброспинальной жидкости.
При заболевании пациентам назначаются шунтирующие операции, в ходе которых дренируется цереброспинальная жидкость из центрального канала спинного мозга, который предварительно расширяется. При люмбоперитонеальном дренировании цереброспинальную жидкость отводят в область грудной или брюшной полости.
Существует несколько способов лечения заболевания. Выбор определенного способа проводится доктором в соответствии с особенностями протекания патологии.
Краниовертебральные аномалии: разновидности
Краниовертебральные мальформации – это врожденные пороки строения области сочленения шейных позвонков с черепом (затылочной костью). Некоторые формы этих пороков вызывают довольно неприятные симптомы, от простого головокружения до инсультов головного и спинного мозга. Симптоматика может долгое время отсутствовать, а затем развиться внезапно, после травмы, гриппа или другой провокации, причем в любом возрасте.
Вот формы пороков, с которыми мы сталкиваемся наиболее часто:
- Аномалия Кимерли – дополнительная костная дужка первого шейного позвонка, может сдавливать позвоночные артерии, питающие головной мозг кровью;
- Аномалия Арнольда-Киари – выпадение части мозжечка в слишком широкое затылочное отверстие;
- Конкресценция шейных позвонков или синдром Клиппеля-Фейля (Клиппель-Фейля) – сращение двух-трех шейных позвонков между собой, часто приводит к сдавлению позвоночных артерий, перегрузке и повреждению выше- и нижележащих межпозвонковых дисков;
- Базиллярная импрессия – “ввернутость” краев затылочного отверстия внутрь полости черепа;
- Платибазия – врожденный дефект затылочной кости, приводит к смещению мозжечка;
- Ассимиляция атланта – приращение 1-го шейного позвонка к затылочной кости, может вызывать сдавление позвоночных артерий при поворотах головы;
- Проатлант – добавочный, часто нестабильный 1-й шейный позвонок, нередко смещается и ущемляет спинной мозг и позвоночные артерии.
В нашей клинике Вы можете получить необходимое нехирургическое лечение, уточнить диагноз, узнать что можно и что нельзя при краниовертебральных мальформациях вообще и конкретно в Вашем случае.
Подробнее о наиболее распространенных краниовертебральных мальформациях читайте ниже.
Общие рекомендации пациентам с краниовертебральными аномалиями
Аномалия Арнольда-Киари:
Избегать ударов и резких движений головой и шеей, форсированных физических нагрузок, стоек на голове;
Ограничить употребление соли, не употреблять воду и влажную пищу, позже, чем за 2-3 часа до сна;
В случае ухудшения состояния (упорная головная боль, тошнота, рвота, нарушения речи, зрительные расстройства, нарушения координации, слабость одной или нескольких конечностей, расстройства чувствительности, нарушения мочеиспускания) – немедленное обращение к врачу;
Осторожность при выполнении массажа и мануальной терапии;
Периодический контрольный осмотр неврологом.
Аномалия Кимерли:
Избегать околопредельных поворотов головой и шеей, ударов головой и шеей, форсированных физических нагрузок, стоек на голове;
В случае ухудшения состояния (головокружение, упорная головная боль, тошнота, рвота, нарушения речи, зрительные расстройства, нарушения координации, слабость одной или нескольких конечностей, расстройства чувствительности, нарушения мочеиспускания) – немедленное обращение к врачу;
Осторожность при выполнении массажа и мануальной терапии;
Периодический контрольный осмотр неврологом.
Аномалия Арнольда-Киари — симптомы
Говоря о симптомах аномалии, надо, прежде всего, различать вариационные ее типы. Первый тип мальформации сопровождается несколькими синдромами, среди которых: гипертензионный, церебеллярный, сирингомиелический, бульбарный и т.д.
Гипертензионный синдром представляет собой повышение давления внутри черепа (ВЧД). Характерными симптомами будут интенсивные затылочные боли, тошнота, рвота, ригидность шейных мышц.
Церебеллярный синдром характеризуется речевыми расстройствами, нарушениями двигательной функции. При этом отсутствует четкость движений, затруднена мелкая моторика.
Сирингомиелический синдром проявляется потерей чувствительности в конечностях. Больной может получить случайный ожог, не заметив этого сразу. При обследовании обнаруживаются кисты спинного мозга.
Другие типы аномалии сопровождаются более тяжелой симптоматикой.
Справочно. У новорожденных страдает дыхание, возможна его остановка, отмечаются нарушения в глотании. Ребенок не может полноценно питаться. Посинение кожных покровов, гипертонус мышц, нистагм — вот основные проявления аномалии этих типов.
Прогноз аномалии Киари
Прогноз для больного с аномалией Киари зависит от того, какой тип патологии у него был обнаружен. Например, больные с аномалией Киари первого типа могут всю жизнь не нуждаться в специальном лечении. Однако, если у пациентов, у которых был диагностирован первый или второй тип заболевания, появились выраженные симптомы, им необходимо срочно пройти осмотр у невролога. Вовремя проведенная операция поможет избежать серьезных осложнений. Аномалия Киари третьего типа практически не поддается лечению и очень часто приводит к смерти пациента.
Заболевание может сопровождаться рядом серьезных осложнений. В частности, у больного может быть диагностировано нарастание внутричерепной гипертензии, а также накопление в полости черепа жидкости (гипертензионно-гидроцефальный синдром). Поскольку некоторые пациенты с аномалией Киари не могут самостоятельно передвигаться, у них вероятно появление застойных пневмоний. В случае сложного течения болезни могут происходить нарушения дыхания и даже его остановка.
Клинические проявления
Аномалия Арнольда Киари 1 степени тяжести проявляется в виде регулярно возникающих симптомов, таких как частые сильные головные боли, головокружения, шум в голове и ушах. Цветные или черно-белые мушки перед глазами. Еще один характерный симптом – боль и ощущение дискомфорта в шейном отделе позвоночника, часто у таких пациентов отмечается низкое артериальное давление, это связанно с воздействием на сосудистые центры. Со временем может возникать нарушение тактильной и тепловой чувствительности, параличи и парезы конечностей, особо остро данный симптом проявляется в период быстрого роста ребенка. Иногда возникают проблемы с дыханием, ребенок требует наблюдения в динамике у невропатолога. Один из ярких симптомов — нарушение речи. При правильно подобранном лечении и регулярных занятиях с логопедом это проявление поддается незначительной коррекции.
При втором типе мальформации Киари симптомы проявляются после рождения ребенка в виде нарушения дыхания, глотания, ребенок пассивный, крик слабый. Таких деток зачастую помещают в отделение интенсивной терапии и реанимации, так как могут возникнуть и другие симптомы нарушения витальных функций. Как правило, в первые дни проводится оперативное лечение, цель — спасти ребенку жизнь, прогноз для социализации неблагоприятен.
При заболеваниях 3 и 4 степеней тяжести у пациентов наблюдается тремор конечностей, потеря сознание, отсутствие крика на первой минуте жизни, значительные нарушения всех видов чувствительности, атаксии различной выраженности, слепота, реже глухота, нарушение тазовых функций, самопроизвольное мочеиспускание, нарушения координации.
Прогноз для продолжительности жизни зависит от сроков, на которых было выявлено заболевание, своевременности лечения и самоотверженности родителей.
Симптоматика, признаки патологии
Болезнь сопровождается специфическими синдромами. Выраженность и количество этих симптомокомплексов зависит от тяжести порока, какие внутричерепные и позвоночные структуры затронуты патологией.
Характеристика неврологических синдромов:
| Название синдрома | Характер проявления | Симптомы аномалии |
|---|---|---|
| Церебральный (мозжечковый) | Расстройство координации движений, неврологические нарушения, неконтролируемый нистагм (непроизвольное подергивание, колебание глаз) | Нет точности мелкой моторики, походка шаткая, неустойчивость вертикального положения тела.
Тремор (дрожание) пальцев, ухудшение речи, двоение картинки (нарушение зрения). У детей отмечается умственная недостаточность |
| Гипертензионно-гидроцефальный | Нарушение оттока и всасывания ликвора, повышенное внутричерепное давление (интракраниальная гипертензия), водянка мозга | Распирающая головная боль.
Тошнота, рвота вне зависимости от приема лекарств, пищи, напряжение мышц на затылке. Ухудшение самочувствия после пробуждения, движения головой. |
| Бульбарно-пирамидный | Сдавливание пирамид, моста, продолговатого мозга | Паралич либо парез (слабость) мышечной группы на руке, туловище.
Боль/дискомфорт в зоне затылка. Усиливается во время чихания, движений головы. Онемение участков кожи, тела. Нарушение глотательной, дыхательной, речевой функции. |
| Вертебробазилярной недостаточности | Сдавливание кровеносных сосудов, ухудшение питания мозга, кислородное голодание тканей (гипоксия) | Приступы головокружения, полуобморочное состояние, потери сознания.
Ухудшение мышечной силы. Нарушения зрения, слуха. |
| Корешковый | Поражение подъязычного, блуждающего, других нервов и ядер в зоне затылочной кости, большого отверстия, атланто-аксиального сустава, мозжечка | Онемение в области лица, языка.
Нарушения глотания, речи, слуха. |
| Сирингомиелитический | Развитие кисты в веществе продолговатого/спинного мозга, сохраняется восприятие тела в пространстве | Пропадает на коже предплечий, рук, туловища чувствительность к раздражителям, смене температуры (нет боли от ожогов, ран, обморожения).
Слабость и атрофия мышц конечностей, туловища, головы, деформация мелких суставов, искривление позвоночника. Возможно недержание кала, мочи, иная дисфункция выделительных органов. |
Признаки 1 типа аномалии
В случае симптомного течения болезни у человека наблюдается до трех синдромов: вертебробазилярной недостаточности, гипертензионно-гидроцефальный и/или мозжечковый. При этой тяжести порока Арнольда-Киари возможны регулярные приступы мигрени, распирающая боль в зоне затылка. Ухудшается четкость движений, мелкой моторики.
Наблюдаются признаки скачков давления и гипоксии:
- головокружение;
- перед глазами мелькание «мушек»;
- шум в ушах;
- слабость.
Человек ощущает дискомфорт сзади у основания черепа, при поворотах/наклонах головы. В случае прогрессирования возможно присоединение других симптомокомплексов. Это проявление бульбарно-пирамидного, корешкового и/или сирингомиелического синдрома.
Признаки 2 степени
При втором типе порока врачи выявляют одновременно минимум три неврологических синдрома. Родившийся ребенок мало двигается, слабо кричит, ручки согнуты, мышцы напряжены. Есть дисфункция нервной системы, органов.
Пороку Арнольда-Киари 2 типа характерно:
- систематическая остановка дыхания;
- ослабление глоточных мышц;
- синюшность кожи.
Осложняется вторая степень частичным либо полным параличом. Утрата двигательных функций отмечается в пальцах, руке/ноге, в области головы, всего тела.
Признаки 3 степени
Третьей степени мальформации Арнольда-Киари характерно проявление 5―6 синдромов одновременно. Ребенок не кричит сразу после рождения. У пациентов часто отмечается потеря слуха, зрения, дисфункция выделительных органов. Нет четкой координации движения, по утрам возникает рвота, есть аномалии строения позвоночника, головы.
Признаки 4 типа
У людей с четвертой степенью тяжести порока преобладает мозжечковый и гипертензионно-гидроцефальный синдром. Дети беспокойны, слабо вскрикивают, тихо плачут. Если выживают, на обследованиях выявляют снижение интеллекта, отсутствие нормального физического развития. В целом, симптомы с прогнозом жизни третьей и четвертой степени тяжести похожи.